第三章 双原子分子结构与性质
 知识点一:H2+ 结构和及其解
知识点一:H2+ 结构和及其解
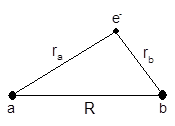
H2+的模型
在波恩- 奥本海默(Born -Oppenheimer)定核近似下, H2+ 体系中电子运动的薛定谔方程为:
![]()
上式中的E是含电子能量和核排斥能在内的H2+分子体系的能量,而波函数则描述了在该确定核构型下电子的可能运动状态。
1.变分原理和线性变分法
求解上述方程需要利用变分原理。变分原理的定义是:若![]() 为给定体系的哈密顿算符,f 为满足体系边界条件的品优函数,则下式成立:
为给定体系的哈密顿算符,f 为满足体系边界条件的品优函数,则下式成立: ![]()
![]()
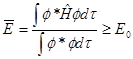
此式称为变分积分。式中f 称为变分函数, E0为体系基态能量,表明体系![]() 关于f 的平均值E存在极小值,这就是变分原理。
关于f 的平均值E存在极小值,这就是变分原理。
当运用变分原理过程中,如果变分函数是采用一组满足体系边界条件且线性无关的品优函数![]() )的线性组合来构造,则被称为线性变分法。即线性变分函数为:
)的线性组合来构造,则被称为线性变分法。即线性变分函数为:
![]()
![]() 是函数
是函数![]() 的线性组合系数。
的线性组合系数。
把变分函数代入变分原理式可得:
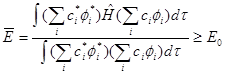
积分后变分积分只剩下![]() 为待定常数。即:
为待定常数。即:
![]()
![]() 在极小值点有:
在极小值点有:
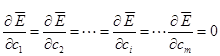
由此可得m 个求解![]() 的联立方程组,称为久期方程。运用线性代数方法可求得久期方程的m套非零解。其中与
的联立方程组,称为久期方程。运用线性代数方法可求得久期方程的m套非零解。其中与![]() 最低值对应的一套
最低值对应的一套![]() 解,组成电子基态近似波函数,该
解,组成电子基态近似波函数,该![]() 便是相应基态能量的近似值。
便是相应基态能量的近似值。
2.分子轨道(MO)和原子轨道线性组合成分子轨(LCAO-MO)
如果由a氢原子和b氢原子的原子轨道![]() 和
和![]() 的线性组合来构造H2+的变分波函数,则
的线性组合来构造H2+的变分波函数,则
![]() (4-1.7)
(4-1.7)
![]() 描述的是分子中单电子的运动状态,称为分子轨道,并把由原子轨道线性组合为分子轨道法简写为LCAO-MO法。
描述的是分子中单电子的运动状态,称为分子轨道,并把由原子轨道线性组合为分子轨道法简写为LCAO-MO法。
3.久期方程组、久期行列式及其解
![]() (4-1.8)
(4-1.8)
这是待定系数c1 、c2的齐次线性方程组,其中
![]() (4-1.9a)
(4-1.9a)
![]() (4-1.9b)
(4-1.9b)
![]() (4-1.9c)
(4-1.9c)
![]() (4-1.9d)
(4-1.9d)
数学上,必有c1= c2=0的一组解,但这组解会导致波函数![]() 为零 ,与H2+有一电子的物理事实相悖;这就需要去求c1 和c2的非零解。
为零 ,与H2+有一电子的物理事实相悖;这就需要去求c1 和c2的非零解。
根据数学相关知识,c1 和c2的非零解的前提条件是下式必须成立:
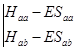
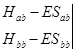 =0 (4-1.10)
=0 (4-1.10)
该式被称为久期行列式。
展开行列式得:
![]() (4-1.14)
(4-1.14)
E有两个解:
![]()
![]() (4-1.15)
(4-1.15)
若![]() 、
、![]() 为H的1s轨道,E1、E2即为H2+ 的基态和第一激发态的能量解。
为H的1s轨道,E1、E2即为H2+ 的基态和第一激发态的能量解。
(d)久期方程的解 把E1代入4-1.12式,可得c1= c2=c',相应的波函数为:
![]() (4-4.16)
(4-4.16)
利用归一化条件有:
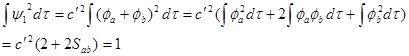
即归一化常数: ![]()
把E2代入4-1.12式,用同样的方法可求得![]() 。
。
这样,H2+基态和第一激发态分子轨道波函数为:
![]()
![]() (4-1.17)
(4-1.17)
4.解的讨论
(a)![]() 和
和![]()
![]() 称为重叠积分(常简写为S),代表了
称为重叠积分(常简写为S),代表了![]() 、
、![]() 在分子中重叠程度。
在分子中重叠程度。
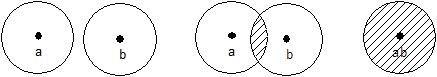
R→∞, Sab=0 R=2a0, Sab=0.586 R=0, Sab=1
(a) (b) (c)
图4-1.2 φa..1s和φb.1s间的重叠
![]() 称为库仑积分(常写为α):
称为库仑积分(常写为α):
![]()
![]()
![]()
![]() (4-1.18)
(4-1.18)
在平衡核间距处J约为![]() 的5%。故
的5%。故![]() 。
。
![]() 称为交换积分(常写为β):
称为交换积分(常写为β):
 (4-1.19)
(4-1.19)
![]()
在平衡核间距处,K<0、![]() >0、
>0、![]() ,故
,故![]() 。
。
(b) E1和E2
![]()
![]()
因E1、E2都是核间距R的函数,能量随R 变化的曲线如图4-1.3。E1—R曲线有能量最低点,此处的R值称为平衡核间距记为Re,对应的能量为H2+的离解能De。该曲线还表明,当H 和H+从无穷远处相互接近时,体系能量逐渐降低,直到R=Re时形成稳定分子。故称与E1对应的态为吸引态、成键态是H2+的基态。而曲线Ⅱ没有极小值,E2随R的增加而单调下降,直到![]() 时
时![]() 。即E2对应的状态始终比原子体系的能量高,此状态下的H2+将自发地离解为原子体系,故被称为排斥态、反键态,是H2+的第一激发态。
。即E2对应的状态始终比原子体系的能量高,此状态下的H2+将自发地离解为原子体系,故被称为排斥态、反键态,是H2+的第一激发态。
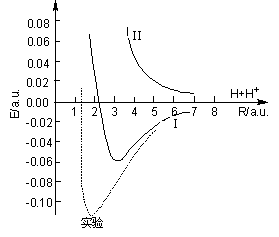
图4-1.3 H2+的能量曲线
(以H+H+体系能量为原点, I 和 II分别为能量E1、E2对R的曲线)
为了简明地表示能量的相对关系,在忽略![]() 后,H2+的能量为:
后,H2+的能量为:
![]() (4-1.20a)
(4-1.20a)
![]() (4-1.20b)
(4-1.20b)
H2+的MO能级图
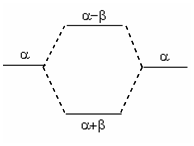
图4-1.4 H2+的MO能级图
(c)![]()
![]()
![]()
![]() 是H2+的吸引态、成键态,当电子排入该轨道时可使分子体系能量降低,称为成键分子轨道。而
是H2+的吸引态、成键态,当电子排入该轨道时可使分子体系能量降低,称为成键分子轨道。而![]() 描述的是排斥态、反键态,当电子排入其中可使分子能量升高,称为反键分子轨道。
描述的是排斥态、反键态,当电子排入其中可使分子能量升高,称为反键分子轨道。
图4-1.5给出了![]() 、
、![]() 的等值线图。在二核之间
的等值线图。在二核之间![]() 有较大几率振幅,没有节面,而
有较大几率振幅,没有节面,而![]() 在核间值则较小且存在节面。从该图还可以看出,分子轨道不是原子轨道电子云的简单的加和,而是发生了波的叠加和强烈的干涉作用。
在核间值则较小且存在节面。从该图还可以看出,分子轨道不是原子轨道电子云的简单的加和,而是发生了波的叠加和强烈的干涉作用。
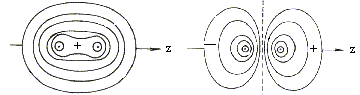
(a) (b)
图4-1.5 H2+ 的![]() (a)和
(a)和![]() (b)的等值线图
(b)的等值线图
(d)共价键本质
H2+成键态电子云图表明,成键后电子云向两核间集中,被形象地称为电子桥。当二个原子相互接近时,由于原子轨道间的叠加,产生强烈的干涉作用,使核间电子密度增大,核间大的电子密度把两核结合到一起,使体系能量降低,形成稳定分子,带来共价键效应。
请同学们继续学习
